


























![String operations
Substrings
>>> #Let’s print only the coding sequence
>>> print(full_seq[3:9])
CATATT
• To understand how we did it we need to
know how strings are numbered:
A T G C A T A T T T G A
0 1 2 3 4 5 6 7 8 9 10 11
Python always starts counting from zero!!!](https://image.slidesharecdn.com/pythonlecturecolor-170530180420/85/Introduction-to-Python-for-Bioinformatics-30-320.jpg)
![String operations
Substrings
>>> #Let’s print only the coding sequence
>>> print(full_seq[3:9])
CATATT
• How to create a substring:
A T G C A T A T T T G A
0 1 2 3 4 5 6 7 8 9 10 11](https://image.slidesharecdn.com/pythonlecturecolor-170530180420/85/Introduction-to-Python-for-Bioinformatics-31-320.jpg)
![String operations
Substrings
>>> #Let’s print only the coding sequence
>>> print(full_seq[3:9])
CATATT
• How to create a substring:
A T G |C A T A T T T G A
0 1 2 [3 4 5 6 7 8 9 10 11
The first number is included (start inclusive).](https://image.slidesharecdn.com/pythonlecturecolor-170530180420/85/Introduction-to-Python-for-Bioinformatics-32-320.jpg)
![String operations
Substrings
>>> #Let’s print only the coding sequence
>>> print(full_seq[3:9])
CATATT
• How to create a substring:
A T G |C A T A T T |T G A
0 1 2 [3 4 5 6 7 8 ]9 10 11
The first number is included (start inclusive).
The second number is excluded (end exclusive).](https://image.slidesharecdn.com/pythonlecturecolor-170530180420/85/Introduction-to-Python-for-Bioinformatics-33-320.jpg)
![String operations
Substrings
>>> #Let’s print only the coding sequence
>>> print(full_seq[3:9])
CATATT
• How to create a substring:
A T G |C A T A T T |T G A
0 1 2 [3 4 5 6 7 8 ]9 10 11
The first number is included (start inclusive).
The second number is excluded (end exclusive).](https://image.slidesharecdn.com/pythonlecturecolor-170530180420/85/Introduction-to-Python-for-Bioinformatics-34-320.jpg)
![String operations
Substrings
>>> #We can also print just one letter
>>> print(full_seq[11])
A
• Each character in the string can be called
using their postion (index) number:
A T G C A T A T T T G A
0 1 2 3 4 5 6 7 8 9 10 11](https://image.slidesharecdn.com/pythonlecturecolor-170530180420/85/Introduction-to-Python-for-Bioinformatics-35-320.jpg)









![Using lists
A list is an object containing several elements:
>>> nucleic_ac = [“DNA”,”mRNA”,”tRNA”]
>>> print(type(nucleic_ac))
<type 'list'>
• A list is created using brackets [ ].
• The elements are separated by commas.
• List elements can be of any object type.](https://image.slidesharecdn.com/pythonlecturecolor-170530180420/85/Introduction-to-Python-for-Bioinformatics-45-320.jpg)
![Using lists
It is possible to mix object types within lists:
>>> number_one = [“one”, 1, 1.0]
>>> numbers_123 = [[“one”, 1, 1.0],
... [“two”, 2, 2.0],[“three”, 3, 3.0]]
We can even make lists of lists!](https://image.slidesharecdn.com/pythonlecturecolor-170530180420/85/Introduction-to-Python-for-Bioinformatics-46-320.jpg)
![Using lists
Elements are called using their index:
>>> number_one = [“one”, 1, 1.0]
>>> numbers_123 = [[“one”, 1, 1.0],
... [“two”, 2, 2.0],[“three”, 3, 3.0]]
>>> print(number_one[1],
... type(number_one[1]))
(1, <type 'int'>)
Don’t forget to start counting from zero!](https://image.slidesharecdn.com/pythonlecturecolor-170530180420/85/Introduction-to-Python-for-Bioinformatics-47-320.jpg)
![Using lists
Elements are called using their index:
>>> number_one = [“one”, 1, 1.0]
>>> numbers_123 = [[“one”, 1, 1.0],
... [“two”, 2, 2.0],[“three”, 3, 3.0]]
>>> print(number_one[2],
... type(number_one[2]))
(1.0, <type ’float'>)](https://image.slidesharecdn.com/pythonlecturecolor-170530180420/85/Introduction-to-Python-for-Bioinformatics-48-320.jpg)
![Using lists
Elements are called using their index:
>>> number_one = [“one”, 1, 1.0]
>>> numbers_123 = [[“one”, 1, 1.0],
... [“two”, 2, 2.0],[“three”, 3, 3.0]]
>>> print(numbers_123[0],
... type(numbers_123[0]))
(['one', 1, 1.0], <type 'list'>)](https://image.slidesharecdn.com/pythonlecturecolor-170530180420/85/Introduction-to-Python-for-Bioinformatics-49-320.jpg)
![Using lists
Elements can be substituted using their index:
>>> numbers_123 = [[“one”, 1, 1.0],
... [“two”, 2, 2.0],[“three”, 3, 3.0]]
>>> numbers_123[0] = [“zero”, 0, 0.0]
>>> print(numbers_123)
[['zero', 0, 0.0], ['two', 2, 2.0],
['three', 3, 3.0]]](https://image.slidesharecdn.com/pythonlecturecolor-170530180420/85/Introduction-to-Python-for-Bioinformatics-50-320.jpg)
![Using lists
The .append() method adds elements to lists:
>>> number_one = [“one”, 1, 1.0]
>>> number_one.append(“I”)
>>> print(number_one)
['one', 1, 1.0, 'I']
• Takes only one of argument.
• Doesn’t return anything, it modifies the
actual list.
• It only adds an element to the end of a list.](https://image.slidesharecdn.com/pythonlecturecolor-170530180420/85/Introduction-to-Python-for-Bioinformatics-51-320.jpg)
![Using lists
Sublists can also be created using indices:
>>> number_one = [“one”, 1, 1.0,”I”]
>>> number_1 = number_one[1:3]
>>> print(number_1, type(number_1))
([1, 1.0], <type 'list'>)
• Work similar to strings (first inclusive,
last exclusive).](https://image.slidesharecdn.com/pythonlecturecolor-170530180420/85/Introduction-to-Python-for-Bioinformatics-52-320.jpg)
![Using loops
Loops make it easier to act on list elements:
>>> nucleic_ac = [“DNA”,“mRNA”,“tRNA”]
>>> for string in nucleic_ac:
... print(string + “ is a nucleic acid”)
...
DNA is a nucleic acid
mRNA is a nucleic acid
tRNA is a nucleic acid](https://image.slidesharecdn.com/pythonlecturecolor-170530180420/85/Introduction-to-Python-for-Bioinformatics-53-320.jpg)
![Using loops
Loops have the following structure:
>>> nucleic_ac = [“DNA”,“mRNA”,“tRNA”]
>>> for string in nucleic_ac:
... print(string + “ is a nucleic acid”)
...
DNA is a nucleic acid
mRNA is a nucleic acid
tRNA is a nucleic acid
• Loop statement:
for ____ in ____ :
Don’t forget the colon!](https://image.slidesharecdn.com/pythonlecturecolor-170530180420/85/Introduction-to-Python-for-Bioinformatics-54-320.jpg)
![Using loops
Loops have the following structure:
>>> nucleic_ac = [“DNA”,“mRNA”,“tRNA”]
>>> for string in nucleic_ac:
... print(string + “ is a nucleic acid”)
...
DNA is a nucleic acid
mRNA is a nucleic acid
tRNA is a nucleic acid
• Element name
• Same rules as variable naming.
This variable only exists inside the loop!](https://image.slidesharecdn.com/pythonlecturecolor-170530180420/85/Introduction-to-Python-for-Bioinformatics-55-320.jpg)
![Using loops
Loops have the following structure:
>>> nucleic_ac = [“DNA”,“mRNA”,“tRNA”]
>>> for acid in nucleic_ac:
... print(acid + “ is a nucleic acid”)
...
DNA is a nucleic acid
mRNA is a nucleic acid
tRNA is a nucleic acid
• Element name
• Same rules as variable naming.
Chose appropriate names to avoid confusion.](https://image.slidesharecdn.com/pythonlecturecolor-170530180420/85/Introduction-to-Python-for-Bioinformatics-56-320.jpg)
![Using loops
Loops have the following structure:
>>> nucleic_ac = [“DNA”,“mRNA”,“tRNA”]
>>> for acid in nucleic_ac:
... print(acid + “ is a nucleic acid”)
...
DNA is a nucleic acid
mRNA is a nucleic acid
tRNA is a nucleic acid
• Iterable object
• The loop elements will depend on the
type of object.](https://image.slidesharecdn.com/pythonlecturecolor-170530180420/85/Introduction-to-Python-for-Bioinformatics-57-320.jpg)

![Using loops
Loops have the following structure:
>>> nucleic_ac = [“DNA”,“mRNA”,“tRNA”]
>>> for acid in nucleic_ac:
... print(acid + “ is a nucleic acid”)
...
DNA is a nucleic acid
mRNA is a nucleic acid
tRNA is a nucleic acid
• The body of the loop is defined with tabs.
• It can be as long as necessary, but all lines
must start with a tab.](https://image.slidesharecdn.com/pythonlecturecolor-170530180420/85/Introduction-to-Python-for-Bioinformatics-59-320.jpg)
![Using loops
Loops have the following structure:
>>> nucleic_ac = [“DNA”,“mRNA”,“tRNA”]
>>> for acid in nucleic_ac:
... print(acid + “ is a nucleic acid”)
... print(“I like “ + acid)
...
DNA is a nucleic acid
I like DNA
mRNA is a nucleic acid
I like mRNA
tRNA is a nucleic acid
I like tRNA](https://image.slidesharecdn.com/pythonlecturecolor-170530180420/85/Introduction-to-Python-for-Bioinformatics-60-320.jpg)
















![Conditions
Let’s evaluate more conditions:
>>> print( len(“ATGC”) > 5 )
False
>>> print( “ATGCGATT”.count(“A”) != 0 )
True
>>> print( “U” in [“A”,”C”,”G”,”T”] )
False
>>> print( “A” in [“A”,”C”,”G”,”T”] )
True
>>> print( len([“A”,”C”,”G”,”T”]) == 4 )
True
>>> print( “ATGCGATT”.isupper())
True
>>> print( “ATGCGATT”.islower())
False](https://image.slidesharecdn.com/pythonlecturecolor-170530180420/85/Introduction-to-Python-for-Bioinformatics-77-320.jpg)

![Conditional tests
An if statement only executes if the condition
evaluates as True:
>>> seq_list = [‘ATTGCATGGTATCTACGG’,
... ‘ATCGCA’,’ATTTTCA’,’ATTCATCGAT’]
>>> for seq in seq_list:
... if len(seq) < 10:
... print(seq)
...
ATCGCA
ATTTTCA
When nesting commands,
be careful with the tabs !](https://image.slidesharecdn.com/pythonlecturecolor-170530180420/85/Introduction-to-Python-for-Bioinformatics-79-320.jpg)
![Conditional tests
An else statement only executes when the if
statement(s) preceding it evaluate as False:
>>> seq_list = [‘ATTGCATGGTATCTACGG’,
... ‘ATCGCA’,’ATTTTCA’,’ATTCATCGAT’]
>>> for seq in seq_list:
... if len(seq) < 10:
... print(seq)
... else:
... print(str(len(seq))+ ‘ base seq’)
...
18 base seq
ATCGCA
ATTTTCA
10 base seq
Remember: else statements
never have conditions!](https://image.slidesharecdn.com/pythonlecturecolor-170530180420/85/Introduction-to-Python-for-Bioinformatics-80-320.jpg)
![Conditional tests
To create if/else blocks with multiple
conditions, we use elif statements:
>>> for seq in seq_list:
... if len(seq) < 10:
... print(seq)
... elif len(seq) == 10:
... print(seq[:5] + ‘...’)
... else:
... print(str(len(seq))+ ‘ base seq’)
...
18 base seq
ATCGCA
ATTTTCA
ATTCA...](https://image.slidesharecdn.com/pythonlecturecolor-170530180420/85/Introduction-to-Python-for-Bioinformatics-81-320.jpg)
![Boolean operators
Boolean operators let us group several
conditions into a single one:
>>> seq_list = [‘ATTGCATGGTATCTACGG’,’AT’,
... ‘ATCGCA’,’ATTCATCGAT’]
>>> for seq in seq_list:
... if len(seq) < 3 or len(seq) > 15:
... print(str(len(seq))+ ‘ base seq’)
... else:
... print(seq)
...
18 base seq
2 base seq
ATCGCA
ATTCATCGAT](https://image.slidesharecdn.com/pythonlecturecolor-170530180420/85/Introduction-to-Python-for-Bioinformatics-82-320.jpg)




































More Related Content
What's hot (20)
Similar to Introduction to Python for Bioinformatics (20)


















Recently uploaded (20)




















Introduction to Python for Bioinformatics
- 1. An introduction to: PythonJosé Héctor Gálvez, M.Sc. Imagesource:www.katie-scott.com
- 2. Scripting languages • Scripting languages are a type of programming language that are interpreted instead of compiled. • They are generally considered high-level and are usually easier to read and learn. • Examples: • Bash (shell scripting) • R (statistical scripting) • Perl (general-purpose scripting) • Python (general-purpose scripting)
- 3. • A popular, open-source, multi-platform, general-purpose scripting language. • Many extensions and libraries for scientific computing. • Current supported versions: 2.7 and 3.5. Install Python on your computer! • Official Python distribution: https://www.python.org/downloads/ • Jupyter (formerly iPython): https://www.continuum.io/downloads
- 4. Learning Goals 1. Understand strings to print and manipulate text 2. Use the open() function to read and write files 3. Understand lists and use loops to go through them 4. Create your own functions 5. Use conditional tests to add more functionality to scripts
- 5. Leaky pipes - A formatting problem Blergh… All my files are messed up! They are in the wrong format! The program I want to use won’t open them! ⎯ Frustrated bioinformatician • We often require code to parse the output of one program and produce another file as input for a specific software. Parse: To analyze a text to extract useful information from it.
- 7. Handling text in Python Printing text to the terminal: >>> print(“Hello world”)
- 8. Handling text in Python Printing text to the terminal: >>> print(“Hello world”) • Python interpreter prompt: >>>
- 9. Handling text in Python Printing text to the terminal: >>> print(“Hello world”) • Python interpreter prompt: >>> • Input: print(“Hello world”)
- 10. Handling text in Python Printing text to the terminal: >>> print(“Hello world”) • Python interpreter prompt: >>> • Input: print(“Hello world”) • Function: print()
- 11. Handling text in Python Printing text to the terminal: >>> print(“Hello world”) • Python interpreter prompt: >>> • Input: print(“Hello world”) • Function: print() • Argument: “Hello world”
- 12. Handling text in Python Printing text to the terminal: >>> print(“Hello world”) Hello world • Python interpreter prompt: >>> • Input: print(“Hello world”) • Function: print() • Argument: “Hello world” • Output: Hello world
- 13. Handling text in Python Printing text to the terminal: >>> print(“Hello world”) Hello world • Python interpreter prompt: >>> • Input: print(“Hello world”) • Function: print() • Argument: “Hello world” • Output: Hello world
- 14. Handling text in Python What happens if we use single quotes? >>> print(‘Hello world’) Hello world We get the same result!!! • In Python single quotes ‘’ and double quotes “” are interchangeable. But, don’t mix them!
- 15. Handling text in Python What happens if we mix quotes? >>> print(‘Hello world”) File "<stdin>", line 1 print('Hello world") ^ SyntaxError: EOL while scanning single- quoted string Whoops!
- 16. Handling text in Python Error messages give us important clues: >>> print(‘Hello world”) File "<stdin>", line 1 print('Hello world") ^ SyntaxError: EOL while scanning single- quoted string
- 17. Handling text in Python Error messages give us important clues: >>> print(‘Hello world”) File "<stdin>", line 1 print('Hello world") ^ SyntaxError: EOL while scanning single- quoted string • File and line containing error.
- 18. Handling text in Python Error messages give us important clues: >>> print(‘Hello world”) File "<stdin>", line 1 print('Hello world") ^ SyntaxError: EOL while scanning single- quoted string • File and line containing error. • Best guess as to where error is found.
- 19. Handling text in Python Error messages give us important clues: >>> print(‘Hello world”) File "<stdin>", line 1 print('Hello world") ^ SyntaxError: EOL while scanning single- quoted string • File and line containing error. • Best guess as to where error is found. • Error type and explanation.
- 20. Handling text in Python We can save strings as variables: >>> #My first variable! >>> dna_seq1 = “ATGTGA”
- 21. Handling text in Python We can save strings as variables: >>> #My first variable! >>> dna_seq1 = “ATGTGA” • A line starting with # is a comment.
- 22. Handling text in Python We can save strings as variables: >>> #My first variable! >>> dna_seq1 = “ATGTGA” • A line starting with # is a comment. • We use the = symbol to assign a variable. • We can re-assign variables as many times as we want. That’s why they’re called variables !
- 23. Handling text in Python We can save strings as variables: >>> #My first variable! >>> dna_seq1 = “ATGTGA” >>> dna_seq1 = “ATGTAA” • A line starting with # is a comment. • We use the = symbol to assign a variable. • We can re-assign variables as many times as we want. That’s why they’re called variables !
- 24. Handling text in Python We can save strings as variables: >>> print(dna_seq1) ATGTAA • Once assigned, the we can use the variable name instead of its content. • Variable names can have letters, numbers, and underscores. • They can’t start with numbers. • They are case-sensitive. Name your variables carefully!
- 25. Handling text in Python Any value between quotes is called a string: >>> type(dna_seq1) <type ‘str’> • Strings (‘str’) are a type of object. • Other types include integers (‘int’), floats (‘float’), lists (‘list’), etc… • Strings are mainly used to manipulate text within Python. Understanding how to use strings is crucial for bioinformatics!
- 26. String operations Concatenation >>> start_codon = ‘ATG’ >>> stop_codon = ‘TGA’ >>> coding_seq = ‘CATATT’ >>> full_seq = start_codon + coding_seq ... + stop_codon >>> print(full_seq) ATGCATATTTGA • To combine strings, we use the + operator
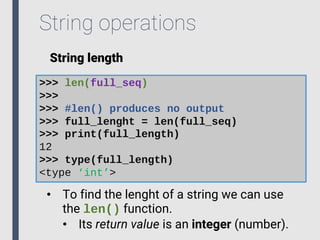
- 27. String operations String length >>> len(full_seq) >>> >>> #len() produces no output >>> full_lenght = len(full_seq) >>> print(full_length) 12 >>> type(full_length) <type ‘int’> • To find the lenght of a string we can use the len() function. • Its return value is an integer (number).
- 28. String operations Turning objects into strings >>> print(“The length of our seq is ” ... + full_length) Traceback (most recent call last): File "<stdin>", line 1, in ? TypeError: cannot concatenate 'str' and 'int' objects • It is not possible to concatenate objects of different types.
- 29. String operations Turning objects into strings >>> print(“The length of our seq is ” ... + str(full_length)) The length of our seq is 12 • The str() function turns any object into a string.
- 30. String operations Substrings >>> #Let’s print only the coding sequence >>> print(full_seq[3:9]) CATATT • To understand how we did it we need to know how strings are numbered: A T G C A T A T T T G A 0 1 2 3 4 5 6 7 8 9 10 11 Python always starts counting from zero!!!
- 31. String operations Substrings >>> #Let’s print only the coding sequence >>> print(full_seq[3:9]) CATATT • How to create a substring: A T G C A T A T T T G A 0 1 2 3 4 5 6 7 8 9 10 11
- 32. String operations Substrings >>> #Let’s print only the coding sequence >>> print(full_seq[3:9]) CATATT • How to create a substring: A T G |C A T A T T T G A 0 1 2 [3 4 5 6 7 8 9 10 11 The first number is included (start inclusive).
- 33. String operations Substrings >>> #Let’s print only the coding sequence >>> print(full_seq[3:9]) CATATT • How to create a substring: A T G |C A T A T T |T G A 0 1 2 [3 4 5 6 7 8 ]9 10 11 The first number is included (start inclusive). The second number is excluded (end exclusive).
- 34. String operations Substrings >>> #Let’s print only the coding sequence >>> print(full_seq[3:9]) CATATT • How to create a substring: A T G |C A T A T T |T G A 0 1 2 [3 4 5 6 7 8 ]9 10 11 The first number is included (start inclusive). The second number is excluded (end exclusive).
- 35. String operations Substrings >>> #We can also print just one letter >>> print(full_seq[11]) A • Each character in the string can be called using their postion (index) number: A T G C A T A T T T G A 0 1 2 3 4 5 6 7 8 9 10 11
- 36. String operations Methods >>> lower_seq = full_seq.lower() >>> print(lower_seq) atgcatatttga • A method is similar to a function, but it is associated to a specific object type. • We call them after a variable of the right type, using a ‘.’ (period) to separate them. • In this case, the method .lower() is called on strings to convert all uppercase characters into lowercase.
- 37. Objective 2: Files in Python Image source: www.katie-scott.com
- 38. Opening files The open() function is used to open files: >>> my_file = open(“BV164695.1.seq”,”r”) >>> print(my_file) <open file ‘BV164695.1.seq', mode 'r' at 0x109de84b0> • It returns a file object. • This object is different from other types of objects. • We rarely interact with it directly. • We mostly interact with it through methods.
- 39. Opening files The open() function is used to open files: >>> my_file = open(“BV164695.1.seq”,”r”) • The first argument is the path to the file. • This path should be relative to our working directory.* • The second argument is the mode in which we are opening the file. • We separate arguments using a comma. Don’t forget the quotes!
- 40. Opening files Files can be opened in three modes: • Read ( “r” ): Permits access to the content of a file, but can’t modify it (default). • Write ( “w” ): Enables the user overwrite the contents of a file. • Append ( “a” ): Enables the user to add content to a file, without erasing previous content. Don’t confuse write and append, you could lose a lot of data!
- 41. Opening files The .read() method extracts file content: >>> my_file = open(“BV164695.1.seq”,”r”) >>> file_content = my_file.read() >>> print(type(my_file), ... type(file_content)) (<type 'file'>, <type 'str'>) • Returns the full contents of a file as a string. • Takes no arguments. Remember: The .read() method can only be used on file objects in read mode!
- 42. Opening files The .write() method writes content into file: >>> out_file = open(“test_out.txt”,”w”) >>> hello_world = “Hello world!” >>> out_file.write(hello_world) • Writes content into file objects in “w” or “a” modes. • Argument must be a string. The .write() method can only be used on file objects in write or append mode!
- 43. Closing files The .close() method flushes a file: >>> print(out_file) <open file ’test_out.txt', mode ’w' at 0x 103f53540> >>> out_file.close() >>> print(out_file) <closed file ’test_out.txt', mode ’w' at 0x103f53540> • Flushing files saves the changes and lets other programs use it. It is always good practice to close files after using them!
- 44. Objective 3: Lists and loops Imagesource:www.katie-scott.com
- 45. Using lists A list is an object containing several elements: >>> nucleic_ac = [“DNA”,”mRNA”,”tRNA”] >>> print(type(nucleic_ac)) <type 'list'> • A list is created using brackets [ ]. • The elements are separated by commas. • List elements can be of any object type.
- 46. Using lists It is possible to mix object types within lists: >>> number_one = [“one”, 1, 1.0] >>> numbers_123 = [[“one”, 1, 1.0], ... [“two”, 2, 2.0],[“three”, 3, 3.0]] We can even make lists of lists!
- 47. Using lists Elements are called using their index: >>> number_one = [“one”, 1, 1.0] >>> numbers_123 = [[“one”, 1, 1.0], ... [“two”, 2, 2.0],[“three”, 3, 3.0]] >>> print(number_one[1], ... type(number_one[1])) (1, <type 'int'>) Don’t forget to start counting from zero!
- 48. Using lists Elements are called using their index: >>> number_one = [“one”, 1, 1.0] >>> numbers_123 = [[“one”, 1, 1.0], ... [“two”, 2, 2.0],[“three”, 3, 3.0]] >>> print(number_one[2], ... type(number_one[2])) (1.0, <type ’float'>)
- 49. Using lists Elements are called using their index: >>> number_one = [“one”, 1, 1.0] >>> numbers_123 = [[“one”, 1, 1.0], ... [“two”, 2, 2.0],[“three”, 3, 3.0]] >>> print(numbers_123[0], ... type(numbers_123[0])) (['one', 1, 1.0], <type 'list'>)
- 50. Using lists Elements can be substituted using their index: >>> numbers_123 = [[“one”, 1, 1.0], ... [“two”, 2, 2.0],[“three”, 3, 3.0]] >>> numbers_123[0] = [“zero”, 0, 0.0] >>> print(numbers_123) [['zero', 0, 0.0], ['two', 2, 2.0], ['three', 3, 3.0]]
- 51. Using lists The .append() method adds elements to lists: >>> number_one = [“one”, 1, 1.0] >>> number_one.append(“I”) >>> print(number_one) ['one', 1, 1.0, 'I'] • Takes only one of argument. • Doesn’t return anything, it modifies the actual list. • It only adds an element to the end of a list.
- 52. Using lists Sublists can also be created using indices: >>> number_one = [“one”, 1, 1.0,”I”] >>> number_1 = number_one[1:3] >>> print(number_1, type(number_1)) ([1, 1.0], <type 'list'>) • Work similar to strings (first inclusive, last exclusive).
- 53. Using loops Loops make it easier to act on list elements: >>> nucleic_ac = [“DNA”,“mRNA”,“tRNA”] >>> for string in nucleic_ac: ... print(string + “ is a nucleic acid”) ... DNA is a nucleic acid mRNA is a nucleic acid tRNA is a nucleic acid
- 54. Using loops Loops have the following structure: >>> nucleic_ac = [“DNA”,“mRNA”,“tRNA”] >>> for string in nucleic_ac: ... print(string + “ is a nucleic acid”) ... DNA is a nucleic acid mRNA is a nucleic acid tRNA is a nucleic acid • Loop statement: for ____ in ____ : Don’t forget the colon!
- 55. Using loops Loops have the following structure: >>> nucleic_ac = [“DNA”,“mRNA”,“tRNA”] >>> for string in nucleic_ac: ... print(string + “ is a nucleic acid”) ... DNA is a nucleic acid mRNA is a nucleic acid tRNA is a nucleic acid • Element name • Same rules as variable naming. This variable only exists inside the loop!
- 56. Using loops Loops have the following structure: >>> nucleic_ac = [“DNA”,“mRNA”,“tRNA”] >>> for acid in nucleic_ac: ... print(acid + “ is a nucleic acid”) ... DNA is a nucleic acid mRNA is a nucleic acid tRNA is a nucleic acid • Element name • Same rules as variable naming. Chose appropriate names to avoid confusion.
- 57. Using loops Loops have the following structure: >>> nucleic_ac = [“DNA”,“mRNA”,“tRNA”] >>> for acid in nucleic_ac: ... print(acid + “ is a nucleic acid”) ... DNA is a nucleic acid mRNA is a nucleic acid tRNA is a nucleic acid • Iterable object • The loop elements will depend on the type of object.
- 58. Using loops Some basic iterable object types: Object type Iterable element List List element String Individual characters Open file in ‘r’ mode Individual line in the file Dictionary Values (in arbitrary order) Set Set element (in arbitrary order) The variety of iterable objects makes loops a very powerful tool in python!
- 59. Using loops Loops have the following structure: >>> nucleic_ac = [“DNA”,“mRNA”,“tRNA”] >>> for acid in nucleic_ac: ... print(acid + “ is a nucleic acid”) ... DNA is a nucleic acid mRNA is a nucleic acid tRNA is a nucleic acid • The body of the loop is defined with tabs. • It can be as long as necessary, but all lines must start with a tab.
- 60. Using loops Loops have the following structure: >>> nucleic_ac = [“DNA”,“mRNA”,“tRNA”] >>> for acid in nucleic_ac: ... print(acid + “ is a nucleic acid”) ... print(“I like “ + acid) ... DNA is a nucleic acid I like DNA mRNA is a nucleic acid I like mRNA tRNA is a nucleic acid I like tRNA
- 61. Objective 4: Functions Image source: www.katie-scott.com
- 62. Creating functions It is possible to create our own functions: >>> def gc_content(seq): ... length = len(seq) ... G_content = seq.count(“G”) ... C_content = seq.count(“C”) ... GC_content =(G_content + C_content) ... / float(length) ... return GC_content ...
- 63. Creating functions Function definitions have this structure: >>> def gc_content(seq): ... length = len(seq) ... G_content = seq.count(“G”) ... C_content = seq.count(“C”) ... GC_content =(G_content + C_content) ... / float(length) ... return GC_content ... • The definition statement def ___________:
- 64. Creating functions >>> def gc_content(seq): ... length = len(seq) ... G_content = seq.count(“G”) ... C_content = seq.count(“C”) ... GC_content =(G_content + C_content) ... / float(length) ... return GC_content ... • The function name • Same naming rules as variables Function definitions have this structure:
- 65. Creating functions >>> def gc_content(seq): ... length = len(seq) ... G_content = seq.count(“G”) ... C_content = seq.count(“C”) ... GC_content =(G_content + C_content) ... / float(length) ... return GC_content ... • The argument(s) of our function • Same naming rules as variables • This part is optional Function definitions have this structure:
- 66. Creating functions >>> def gc_content(seq): ... length = len(seq) ... G_content = seq.count(“G”) ... C_content = seq.count(“C”) ... GC_content =(G_content + C_content) ... / float(length) ... return GC_content ... • The body of the function is defined by tabs • It can be as long as necessary, but all lines must start with a tab. Function definitions have this structure:
- 67. Creating functions >>> def gc_content(seq): ... length = len(seq) ... G_content = seq.count(“G”) ... C_content = seq.count(“C”) ... GC_content =(G_content + C_content) ... / float(length) ... return GC_content ... • The return statement (optional) • Can return one or more objects • Marks the end of a function Function definitions have this structure:
- 68. Calling functions >>> test_seq = “ACTGATCGATCG” >>> gc_test = gc_content(test_seq) >>> print(gc_test, type(gc_test)) (0.5, <type 'float'>) >>> print(GC_content) Traceback (most recent call last): File "<stdin>", line 1, in <module> NameError: name 'GC_content' is not defined Once defined, we can call a function: Variables within the function are not defined outside of that function!
- 69. Other function options >>> test_seq = “ACTGATCGATCG” >>> print(gc_content(test_seq)) 0.5 >>> test_seq = “ACTGATCGATCGC” >>> print(gc_content(test_seq)) 0.538461538462 Let’s improve our function: I don’t want that many numbers!
- 70. Other function options The round() function lets us round the result: >>> def gc_content(seq): ... length = len(seq) ... G_content = seq.count(“G”) ... C_content = seq.count(“C”) ... GC_content =(G_content + C_content) ... / float(length) ... return round(GC_content,2) ... >>> print(gc_content(test_seq)) 0.54
- 71. Other function options A second argument gives more flexibility: >>> def gc_content(seq,sig_fig): ... length = len(seq) ... G_content = seq.count(“G”) ... C_content = seq.count(“C”) ... GC_content =(G_content + C_content) ... / float(length) ... return round(GC_content,sig_fig) ... >>> print(gc_content(test_seq,2)) 0.54 >>> print(gc_content(test_seq,3)) 0.538
- 72. Other function options We can call a function with keyword arguments: >>> def gc_content(seq,sig_fig): ... length = len(seq) ... G_content = seq.count(“G”) ... C_content = seq.count(“C”) ... GC_content =(G_content + C_content) ... / float(length) ... return round(GC_content,sig_fig) ... >>> print(gc_content(seq=‘ACGC’,sig_fig=1)) 0.8 >>> print(gc_content(sig_fig=1,seq=‘ACGC’)) 0.8
- 73. Other function options We can give our functions default values: >>> def gc_content(seq,sig_fig=2): ... length = len(seq) ... G_content = seq.count(“G”) ... C_content = seq.count(“C”) ... GC_content =(G_content + C_content) ... / float(length) ... return round(GC_content,sig_fig) ... >>> print(gc_content(test_seq)) 0.54 >>> print(gc_content(test_seq,sig_fig=3)) 0.538
- 75. Conditions Conditions are pieces of code that can only produce one of two answers: - True - False When required, python tests (or evaluates) the condition and produces the result. >>> print( 3 == 5 ) False >>> print( 3 < 5 ) True >>> print( 3 >= 5 ) False These are not strings!
- 76. Conditions The following symbols are used to construct conditions: Symbol Meaning == Equals > < Greater than, less than >= <= Greater and less than, or equal to != Not equal in Is a value in a list is Are the same object* Remember to use two equals signs when writing conditions!
- 77. Conditions Let’s evaluate more conditions: >>> print( len(“ATGC”) > 5 ) False >>> print( “ATGCGATT”.count(“A”) != 0 ) True >>> print( “U” in [“A”,”C”,”G”,”T”] ) False >>> print( “A” in [“A”,”C”,”G”,”T”] ) True >>> print( len([“A”,”C”,”G”,”T”]) == 4 ) True >>> print( “ATGCGATT”.isupper()) True >>> print( “ATGCGATT”.islower()) False
- 78. Conditional tests An if statement only executes if the condition evaluates as True: >>> test_seq = ‘ATTGCATGGTATCTACGG’ >>> if len(test_seq) < 10: ... print(seq) ... >>> >>> test_seq = ‘ATTGCATGG’ >>> if len(test_seq) < 10: ... print(seq) ... ATTGCATGG • If statements have similar structure to loops
- 79. Conditional tests An if statement only executes if the condition evaluates as True: >>> seq_list = [‘ATTGCATGGTATCTACGG’, ... ‘ATCGCA’,’ATTTTCA’,’ATTCATCGAT’] >>> for seq in seq_list: ... if len(seq) < 10: ... print(seq) ... ATCGCA ATTTTCA When nesting commands, be careful with the tabs !
- 80. Conditional tests An else statement only executes when the if statement(s) preceding it evaluate as False: >>> seq_list = [‘ATTGCATGGTATCTACGG’, ... ‘ATCGCA’,’ATTTTCA’,’ATTCATCGAT’] >>> for seq in seq_list: ... if len(seq) < 10: ... print(seq) ... else: ... print(str(len(seq))+ ‘ base seq’) ... 18 base seq ATCGCA ATTTTCA 10 base seq Remember: else statements never have conditions!
- 81. Conditional tests To create if/else blocks with multiple conditions, we use elif statements: >>> for seq in seq_list: ... if len(seq) < 10: ... print(seq) ... elif len(seq) == 10: ... print(seq[:5] + ‘...’) ... else: ... print(str(len(seq))+ ‘ base seq’) ... 18 base seq ATCGCA ATTTTCA ATTCA...
- 82. Boolean operators Boolean operators let us group several conditions into a single one: >>> seq_list = [‘ATTGCATGGTATCTACGG’,’AT’, ... ‘ATCGCA’,’ATTCATCGAT’] >>> for seq in seq_list: ... if len(seq) < 3 or len(seq) > 15: ... print(str(len(seq))+ ‘ base seq’) ... else: ... print(seq) ... 18 base seq 2 base seq ATCGCA ATTCATCGAT
- 83. Boolean operators There are three boolean operators in python: Boolean operator Boolean operation Result and False and False False True and True True True and False False or False or False False True or True True True or False True not not True False not False True
- 84. True/False functions Functions can return True or False: >>> def is_long(seq,min_len=10): ... if len(seq) > min_len: ... return True ... else: ... return False ... >>> for seq in seq_list: ... if is_long(seq): ... print(‘Long sequence’) ... else: ... print(‘Short sequence’) ...
- 85. True/False functions Functions can return True or False: >>> for seq in seq_list: ... if is_long(seq): ... print(‘Long sequence’) ... else: ... print(‘Short sequence’) ... Long sequence Short sequence Short sequence Short sequence
- 86. True/False functions Functions can return True or False: >>> for seq in seq_list: ... if is_long(seq,5): ... print(‘Long sequence’) ... else: ... print(‘Short sequence’) ... Long sequence Short sequence Long sequence Long sequence
- 87. Conclusion • Python is a very powerful language that is currently used for many things: • Bioinformatics tool development • Pipeline deployment • Big Data analysis • Scientific computing • Web development (Django) The best way to learn to code is through practice and by reading other developers’ code!
- 88. References & Further Reading • Official python documentation: https://www.python.org/doc/ • “Python for Biologists” by Dr. Martin Jones www.pythonforbiologists.com • E-books with biological focus • CodeSkulptor: http://www.codeskulptor.org/ • Codecademy python course: https://www.codecademy.com/learn/python • Jupyter project: http://jupyter.org/index.html