


































More Related Content











































Similar to stability tests for pharmaceutical products (20)








































stability tests for pharmaceutical products
- 1. Prepared By : Ala’a R. alfayez Zainab al-mulla
- 2. Stability Definition These studies provide information about the packaging in that it is not reactive, additive, or absorptive so that the identity, strength, quality and purity of the drug product is not affected, also to provide clearance on stability process flow. To assessment of the stability characteristics of all drug products manufactured / packaged and/or repacked by Pharmaceuticals companies. To establish and extend the shelf life for products
- 3. General Principles The purpose of stability testing is to provide evidence on how the quality of an active substance or pharmaceutical product varies with time under the influence of a variety of environmental factors such as temperature, humidity, and light. In addition, product-related factors influence the stability, e.g. the chemical and physical properties of the active substance and the pharmaceutical excipients, the dosage form and its composition, the manufacturing process, the nature of the container-closure system, and the properties of the packaging materials. Also, the stability of excipients that may contain or form reactive degradation products, have to be considered.
- 4. Active Substance Information on the stability of the active substance is an integral part of the systematic approach to stability evaluation. For active substances not described in an official pharmacopoeial monograph, stability studies are required. For active substances described in an official pharmacopoeial monograph, which covers the degradation products and for which suitable limits have been set but a re-test period is not defined, two options are acceptable:
- 5. Active Substance The manufacturer of the pharmaceutical product confirms that the active substance complies with the pharmacopoeial monograph immediately prior to the manufacture of the pharmaceutical product. In this case no stability studies on the active substance are required. The suitability of the pharmacopoeial monograph for the active substance used from a named source of supply has to be demonstrated.
- 6. Stress Testing Stress testing of the active substance can help identify the likely degradation products, which can in turn help establish the degradation pathways and the intrinsic stability of the molecule and validate the stability indicating power of the analytical procedures used. The nature of the stress testing will depend on the individual active substance and the type of pharmaceutical product involved.
- 7. Stress Testing For an active substance the following approaches may be used: a) When an active substance is described in an official pharmacopoeial monograph, and fully meets its requirements, no data are required on the degradation products if they are named under the headings “purity tests” and/or “section on impurities
- 8. Stress Testing b) For active substances not described in an official pharmacopoeial monograph, there are two options: − When available, it is acceptable to provide the relevant data published in the literature to support the proposed degradation pathways; − When no data are available in the scientific literature, including official pharmacopoeias, stress testing should be performed.
- 9. Specification Stability studies should include testing of those attributes of the active substance that are susceptible to change during storage and are likely to influence quality, safety, and/or efficacy. The testing should cover, as appropriate, the physical, chemical, biological, and microbiological attributes. Validated stability-indicating analytical procedures should be applied. Whether and to what extent replication should be performed will depend on the results from validation studies
- 10. Storage Conditions In general, an active substance should be evaluated under storage conditions (with appropriate tolerances) that test its thermal stability and, if applicable, its sensitivity to moisture. The storage conditions and the lengths of studies chosen should be sufficient to cover storage, shipment, and subsequent use with due regard to the climatic zone(s) in which the active substance is intended to be stored .
- 11. Storage Conditions Station 1: (40 °C 2 & 75 % RH 5). Station 2: (30 °C Station 3: (25 °C 2 & 65 % RH 2 & 60 % RH 5). 5). Station 5: (30 °C Station 4: (2 - 8) °C. 2 & 35 % RH 5).
- 12. Product storage Storage condition Testing stations Stability condition Zone Station conditions Temperature/Humidity (Month) all Room temperature 1 40°C 2°C / 75% 5%RH Accelerated 0, 1, 2,3 and 6 all Refrigerator storage 3 25°C 2°C / 60% 5 %RH 30°C 2°C / 65% 5%RH III, IV Room temperature 2 30°C 2°C / 35% 5%RH 0, 3, 6, 9, 12, Long-Term 18, 24, 36, 48 and 60 I, II Room temperature 3 25°C 2°C / 60% 5 %RH all Refrigerator 4 (2 – 8)°C Intermediate I, II Room temperature 2 30°C 2°C / 65% 5%RH 0, 3, 6, 9 and 12
- 13. Testing Frequency For long term studies, frequency of testing should be sufficient to establish the stability profile of the active substance. For active substances with a proposed re-test period of at least 12 months, the frequency of testing at the long term storage condition should normally be every three months over the first year, every six months over the second year, and annually thereafter through the proposed re-test period.
- 14. Testing Frequency At the accelerated storage condition, a minimum of three time points, including the initial and final time points (e.g. 0, 3, and 6 months), from a 6- month study is recommended. Where an expectation (based on development experience) exists that results from accelerated studies are likely to approach significant change criteria, increased testing should be conducted either by adding samples at the final time point or by including a fourth time point in the study design
- 15. For products stored at room temperature. For products stored at refrigerator. for products stored at room temperature (for long term conditions at 25 °C 60 % RH). For products stored at room temperature in climatic zone III and IV. For products stored at room temperature in climatic zone I and II. For products stored at refrigerator.
- 16. Type of Placement Batches: A- New product registration. B- Process Validation Batch. C- Formulation Change. D- Annual Commitment Batch. E- Raw material manufacturer change F- Primary packaging material manufacturer change G- Reprocessing batch
- 17. H- Batch size change I- Manufacturing process change J- Stability condition change K- Machine change L- Site transfer
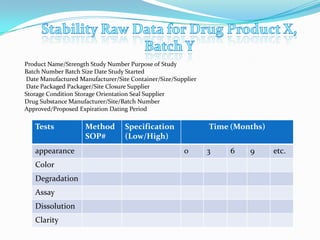
- 18. Product Name/Strength Study Number Purpose of Study Batch Number Batch Size Date Study Started Date Manufactured Manufacturer/Site Container/Size/Supplier Date Packaged Packager/Site Closure Supplier Storage Condition Storage Orientation Seal Supplier Drug Substance Manufacturer/Site/Batch Number Approved/Proposed Expiration Dating Period Tests Method Specification Time (Months) SOP# (Low/High) appearance 0 3 6 9 etc. Color Degradation Assay Dissolution Clarity
- 19. In-use stability The purpose of in-use stability testing is to establish – where applicable – the period of time during which a multi-dose product can be used whilst retaining acceptable quality once the container is opened and the first dose is removed. A minimum of two batches, at least pilot scale batches, should be subjected to the test. At least one of the batches should be chosen towards the end of its shelf life. If such results are not available, one batch should be tested at the final point of the submitted stability studies.
- 20. Variations Once the pharmaceutical product has been registered, additional stability studies are required whenever major variations are made like the following: 1. Change in the manufacturing process; 2. Change in the composition of the pharmaceutical product; 3. Change of the immediate packaging.
- 21. Stability tests for dosage forms: 1. Tablets Dissolution (or disintegration, if justified), water content and hardness/friability. For coated and colour tablets additional tests may require for texture and colour stability. 2. Capsules Hard gelatin capsules: brittleness, dissolution (or disintegration, if justified), water content, and level of microbial contamination. 3. Emulsions Phase separation, pH, viscosity, level of microbial contamination, and mean size and distribution of dispersed globules. 4. Oral solutions and suspensions Formation of precipitate, clarity for solutions, pH, viscosity, extractables, level of microbial contamination.
- 22. Stability tests for dosage forms: Additionally for suspensions, redispersibility, rheological properties, mean size and distribution of particles should be considered. Also, polymorphic conversion may be examined, if applicable. 5. Powders and granules for oral solution or suspension Water content, and reconstitution time. Reconstituted products (solutions and suspensions) should be evaluated as described in “Oral solutions and suspensions” above, after preparation according to the recommended labeling, through the maximum intended use period.
- 23. Stability tests for dosage forms: 6. Nasal sprays: solutions and suspensions Clarity (for solution), level of microbial contamination, pH, particulate matter, unit spray medication content uniformity, number of actuations meeting unit spray content uniformity per container, droplet and/or particle size distribution, weight loss, pump delivery, microscopic evaluation (for suspensions), foreign particulate matter and extractable/leachable from plastic and elastomeric components of the container, closure and pump. 7. Topical, ophthalmic and otic preparations Included in this broad category are ointments, creams, lotions, paste, gel, solutions, eye drops, and cutaneous sprays.
- 24. Stability tests for dosage forms: Topical preparations should be evaluated for clarity, homogeneity, pH, resuspendability (for lotions), consistency, viscosity, particle size distribution (for suspensions, when feasible), level of microbial contamination/sterility and weight loss (when appropriate). Evaluation of ophthalmic or otic products (e.g. creams, ointments, solutions and suspensions) should include the following additional attributes: sterility, particulate matter and extractable. Evaluation of cutaneous sprays should include: pressure, weight loss, net weight dispensed, delivery rate, level of microbial contamination, spray pattern, water content, and particle size distribution (for suspensions). 8. Suppositories Softening range, dissolution (at 37°C).
- 25. Stability tests for dosage forms: 9. Small volume parenterals (SVPs) Colour, clarity (for solutions), particulate matter, pH, sterility, endotoxins. Stability studies for powders for injection solution should include monitoring for colour, reconstitution time and water content. Specific parameters to be examined at appropriate intervals throughout the maximum intended use period of the reconstituted drug product, stored under condition(s) recommended in labelling, should include clarity, colour, pH, sterility, pyrogen/endotoxin and particulate matter. The stability studies for Suspension for injection should include, in addition, particle size distribution, redispersibility and rheological properties. The stability studies for Emulsion for injection should include, in addition, phase separation,viscosity, mean size and distribution of dispersed phase globules.
- 26. Stability tests for dosage forms: 10. Large volume parenterals (LVPs) Colour, clarity, particulate matter, pH, sterility, pyrogen/endotoxin, and volume.
- 27. When re-test is done?? In general, “significant change” for a pharmaceutical product is defined as: 1. A 5% change in assay from its initial value; or failure to meet the acceptance criteria for potency when using biological or immunological procedures; 2. Any degradation product exceeding its acceptance criterion; 3. Failure to meet the acceptance criteria for appearance, physical attributes, and functionality test (e.g., colour, phase separation, resuspendibility, caking, hardness, dose delivery per actuation); however, some changes in physical attributes (e.g., softening of suppositories, melting of creams, partial loss of adhesion for transdermal products
- 28. When re-test is done? 4. Failure to meet the acceptance criterion for pH; or 5. Failure to meet the acceptance criteria for dissolution for 12 dosage units.
- 29. Ongoing stability studies The stability of the API should be monitored according to a continuous and appropriate program that will permit the detection of any stability issue (e.g. changes in levels of degradation products). The purpose of the ongoing stability program is to monitor the API and to determine that the API remains, and can be expected to remain, within specifications under the storage conditions indicated on the label, within the re-test period in all future batches. The ongoing stability program should be described in a written protocol and the results presented in a formal report.
- 31. To support a shelf life extension, a report must be generated documenting stability data (for the proposed interval) for three batches which meet all the following criteria: Same product / potency Same manufacturing Shelf life may be extended as long process. term data become available to justify the extension Same primary packaging material Same formulation No significant change in the manufacturing procedure
- 32. After registration… Once the pharmaceutical product has been registered, additional stability studies are required whenever variations that may affect the stability of the active pharmaceutical substance or pharmaceutical product are made, such as major variations like the following: a. Change in the manufacturing process. b. Change in the composition of the pharmaceutical product. c. Change of the immediate packaging.
- 33. At the market… If no significant change occurs during six-month's accelerated and real time stability testing, the product will be allowed to place in the market with a provisional shelf-life of up to twenty-four months. However, real time stability testing should be continued up to the proposed shelf-life. The manufacturer should have a system of recall in place so that the sale any batch which does not remain within the limit of approved product specification be stopped within twenty-four hours.